
I might return to this later and have a look at how to make the parallel executable in the bin/LINUX64 folder.
Original post:
This is another addition to my growing list over unsuccessful, abandoned or only partially successful builds.
(see e.g.
http://verahill.blogspot.com.au/2013/05/409-failed-attempt-at-compiling-gamess_10.html
http://verahill.blogspot.com.au/2013/05/409a-failed-attempt-at-compiling-gamess.html
http://verahill.blogspot.com.au/2012/08/compiling-dalton-qm-on-debian-in.html
http://verahill.blogspot.com.au/2012/07/quantum-espresso-on-rocks-543-centos-56.html)
In other words -- yes, it builds. But no, it is unusable.
I can build nwchem with openmp support, and it does run in parallel -- but the wall time is enormous since most of the time only a single thread is running.
Maybe someone will read this and see what's missing, or feel inspired to make their own attempt
What I did
ACML libraries were installed as shown in e.g. http://verahill.blogspot.com.au/2013/05/409-failed-attempt-at-compiling-gamess_10.html
Nwchem was downloaded:
sudo mkdir /opt/nwchem sudo chown $USER:$USER /opt/nwchem cd /opt/nwchem wget http://www.nwchem-sw.org/download.php?f=Nwchem-6.1.1-src.2012-06-27.tar.gz tar xvf Nwchem-6.1.1-src.2012-06-27.tar.gz cd nwchem-6.1.1-src/
Next I edited src/config/makefile.h
I then built using the following build script:2363 ifdef OPTIMIZE 2364 FFLAGS = $(FOPTIONS) $(FOPTIMIZE) 2365 CFLAGS = $(COPTIONS) $(COPTIMIZE) -fopenmp 2366 else 2367 # Need FDEBUG after FOPTIONS on SOLARIS to correctly override optimization 2368 FFLAGS = $(FOPTIONS) $(FDEBUG) 2369 CFLAGS = $(COPTIONS) $(CDEBUG) -fopenmp 2370 endif 2371 INCLUDES = -I. $(LIB_INCLUDES) -I$(INCDIR) $(INCPATH) 2372 CPPFLAGS = $(INCLUDES) $(DEFINES) $(LIB_DEFINES) 2373 LDFLAGS = $(LDOPTIONS) -L$(LIBDIR) $(LIBPATH) 2374 LIBS = $(NW_MODULE_LIBS) $(CORE_LIBS) -lgomp 2375
So far so good.export LARGE_FILES=TRUE export TCGRSH=/usr/bin/ssh export NWCHEM_TOP=`pwd` export NWCHEM_TARGET=LINUX64 export NWCHEM_MODULES="all" export PYTHONVERSION=2.7 export PYTHONHOME=/usr export BLASOPT="-L/opt/acml/acml5.3.1/gfortran64_fma4_mp_int64/lib -lacml_mp -lpthread" export USE_OPENMP=y export LIBRARY_PATH="$LIBRARY_PATH:/opt/acml/acml5.3.1/gfortran64_fma4_mp_int64/lib" cd $NWCHEM_TOP/src make clean make nwchem_config make FC=gfortran 2> make.err 1>make.log cd $NWCHEM_TOP/contrib export FC=gfortran ./getmem.nwchem
Where it fails
A picture is probably in order:
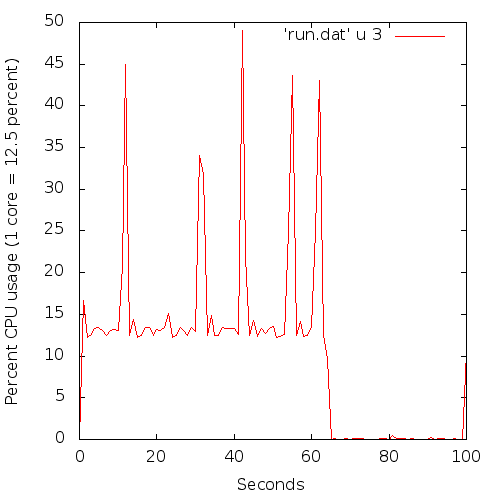
Basically, most of the time only one core is running at 100% (i.e. showing as 12.5 % here since I have 8 cores), with the other cores occasionally kicking in (the 'spikes').
The wall times is 63 seconds, and the 'cpu time' is 83.1 seconds. Ideally, for a fully parallel run the cpu time should be as close to the wall time multiplied with eight for a shared run like this (but is always smaller).
As a comparison, here's an mpi-enabled binary:

So it's not particularly 'parallel' in the OMP case -- but I don't know why. Maybe nwchem 6.1.1 isn't quite ready for OMP yet? I've noticed that it's one of the areas where the upcoming release is supposed to have been improved.
'profiling' with sar -- how-to
sudo apt-get install syssstat
Edit /etc/default/sysstat:
8 # will be overwritten by debconf! 9 ENABLED="true" 10
sudo service sysstat restart
Before launching the run, set sar to run in another windows and collect data before immediately launching the run you want to monitor in a different window:
sar 1 180 >> run.logcollects data every 1 seconds and repeats it 180 times (i.e. 181 seconds) and stores the data in run.log.
No comments:
Post a Comment