I've been using Wsearch (
http://www.wsearch.com.au/wsearch32/wsearch32.htm) to process agilent chemstation ESI-MS spectra for the past few years. It and
Matt Monroe's Molecular Weight calculator (why, oh why is there no comparable molecular weight calculator for linux?) have been the only two reasons why I've bother with Wine under Linux.
Openchrom is written in java and so will run on both Good (Linux) and Evil (OS X and Win) operating systems.
Having finally discovered
openchrom (v 0.6 so still early days) I can now finally retire wsearch from my own computers (it's still a good piece of software, but it's crippled to encourage the purchasing of a 'full' version, and I've had no luck purchasing a license from the author in spite of having tried several times during the past couple of years).
OpenChrom can export an entire agilent experiment as a '3D' csv file which makes processing a lot more fun.
As an aside: I hate proprietary file formats since they prevent me from using my own tools (cat, sed, gawk, gnuplot, octave) when processing -- or at a minimum make it more difficult. Most universities and grant agencies now add a provision regarding data management in their grant acceptance agreements/work conduct policies. In general these provisions state that the data shall be made available publicly and /or managed by a university repository. What is REALLY missing is a clause about using open formats -- and that should be taken into account when acquiring new instrumentation. All else being equal, an instrument which is 'open' will be a lot cheaper to manage in the long run since you won't have to feel locked in in terms of software. That's incidentally a reason why I like Metrohm since they provide details of their RS-232 interface allowing you to write your own software.
Anyway, here's how to get set up:
1. Install Java v1.7 (need > 1.6)
You can either use openjdk 7 or (Oracle) Java. See
here for a general guide to installing Oracle/Sun Java.
As for
openjdk, you can
easily install it:
sudo apt-get install openjdk-7-jdk
(the openjdk-7-jre package is enough if you don't want the full developer's kit)
Anyway.
Make sure that you've selected the right version:
sudo update-alternatives --config java
There are 7 choices for the alternative java (providing /usr/bin/java).
Selection Path Priority Status
------------------------------------------------------------
0 /usr/lib/jvm/java-6-openjdk-amd64/jre/bin/java 1061 auto mode
1 /usr/bin/gij-4.4 1044 manual mode
2 /usr/bin/gij-4.6 1046 manual mode
3 /usr/bin/gij-4.7 1047 manual mode
4 /usr/lib/jvm/j2re1.6-oracle/bin/java 314 manual mode
5 /usr/lib/jvm/j2sdk1.6-oracle/jre/bin/java 315 manual mode
6 /usr/lib/jvm/java-6-openjdk-amd64/jre/bin/java 1061 manual mode
*7 /usr/lib/jvm/java-7-openjdk-amd64/jre/bin/java 1051 manual mode
2. Get openchrom
cd ~/tmp
wget http://sourceforge.net/projects/openchrom/files/REL-0.6.0/openchrom_linux.gtk.x86_64_0.6.0.zip
unzip openchrom_linux.gtk.x86_64_0.6.0.zip
cd linux.gtk.x86_64/OpenChrom/
sudo mkdir /opt/openchrom
sudo chown $USER /opt/openchrom
cp * -R /opt/openchrom
chmod +x /opt/openchrom/openchrom
Stick
alias openchrom='/opt/openchrom/openchrom'
in your ~/.bashrc and source it.
3. Get plugins
On first boot you're asked whether you want to get additional plugins using the '
Openchrom marketplace'.
Since I'm mainly processing data from an Agilent ESI-MS, I wanted the plugin for Agilent files. The website says that you need a license key for plugins BUT that it's free to register for one.
This is a 30-days trial version. Afterwards, you need a valid serial key.
You can get a free serial key after registration on http://www.openchrom.net.
You can use the converter for commercial or non-commercial purposes free of charge, but you are not allowed to redistribute this software without my permission.
Note, that clicking on links on the website didn't lead me to a link to download the plugin. Instead, in OpenChrom click on the
Plug-ins menu:
 |
| As always, make sure you trust your suppliers. |
And then you're done installing.
There's nothing odd about registering other than this: you will receive an email with a confirmation of your registration in clear text WITH YOUR PASSWORD. So...be aware of that.
4. You can now browse in the tree to the left and select your .D folder:
There's a bit of clever thinking when it comes to the functionality of the program. The upside of this I think will eventually be that it's easy to get a consistent experience for a set of users (not unimportant for a research group). The downside is that it's a bit clunky getting started. Play with it for an hour and you'll get the hang of it, so it's not really that much of a hurdle. Also, too many options seem to be context sensitive -- I am having real trouble finding various options under the 'Accurate' perspective which I can find under the 'default' perspective.
5. Some comments:
It's still early days for OpenChrom (v 0.6) , and there are a few minor issues which may or may not affect you:
*
Registration keys. They are easy enough to get (register online, log in, click on the plug in online that you want and you'll see the key), but if you have installed a new plug in and open your first spectrum right after that you'll be asked for registration keys. It won't tell you for which plug in the dialogue you're seeing is though, so if you've just installed three different plug ins you'll have to do some trial-and-error.
This is fixed in the upcoming version.
*
Raw/gaussian plot of mass spectrum. This took a while to figure out, but you have to use perspectives. The default (heavily zoomed in) view looks like this:
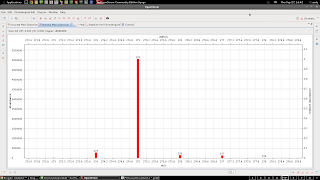 |
| This might be good enough for those organic types...us 108 element inorganic types want more detail |
If you go to the top right corner, click on 'other' (perspectives)
and select accurate you get
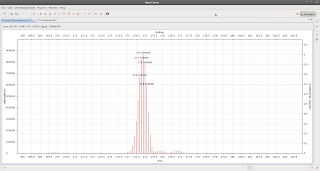 |
| Bingo! |
And then there's the obligatory wish list:
*
A good quality isotopic pattern calculator would be nice. Anyone who has compared the output from different pieces of software will have discovered that different calculators may yield very different patterns. I think some of it boils down to truncation rather than incorrect isotopic ratios, but that just highlights how difficult it can be to implement a seemingly simple concept. The only calculator which I trust AND find useful is Matt Monroe's calculator -- the predicted patterns look good, and you get proper Gaussian broadening which means that it looks 'right'.
This would be perfect as a plug-in. If only I knew how to properly implement it...
*
A good quality ion generator -- some pieces of software (Hi Matt) allow you to select a handful of elements or fragments, pick a range of charges, input an m/z value and based on that spits out a list over possible identities for your signal. It's a good thing to have by your side the first time you look at a complex mixture trying to figure out what products may be present.
This would be perfect as a plug-in. I've written this type of programmes before, but in python using for-loops...a vectorized version should be faster and maybe even easier to write.





















